
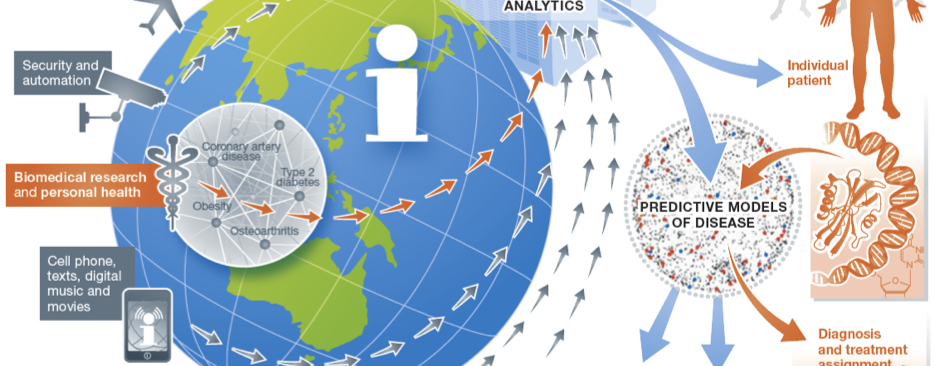
Our Research
Our research focuses on developing and applying translational and biomedical informatics approaches to harness the digital universe of information towards solving critical challenges in precision medicine and enabling a learning healthcare system.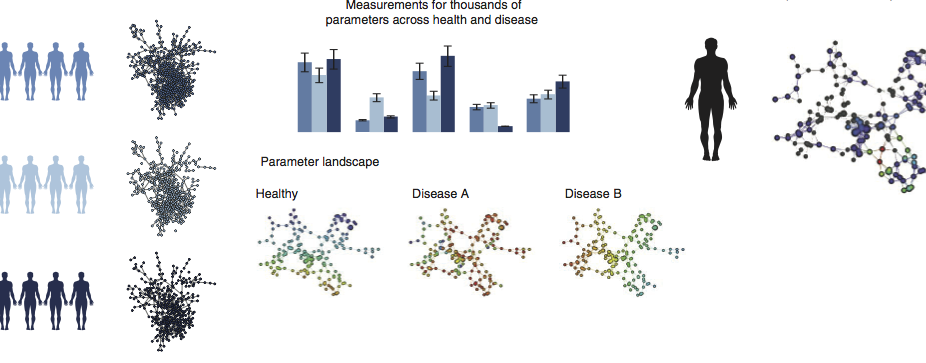
Multiscale Network Modeling of the Immune Systems
Our lab is developing multiscale network-based approaches to better model and investigate the function and dynamics of immune function across cells, tissues, and broader physiology as well as immune response to environmental stimulus and drug therapies.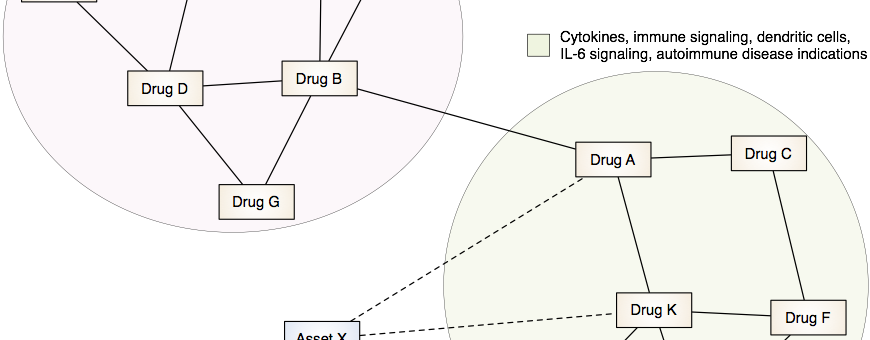
Drug Discovery and Repurposing
Our lab is developing and applying integrative computational approaches to discover novel therapeutics and to identify opportunities to repurpose or reposition drugs to address unmet clinical needs.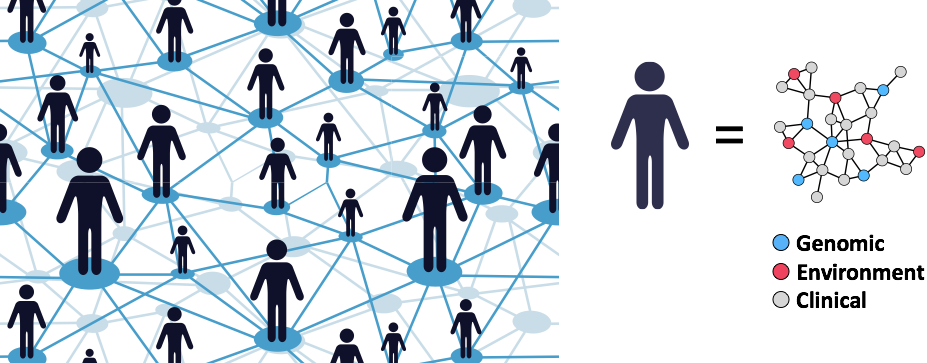
Redefining Human Disease
Our lab is developing data-driven approaches to redefine human disease and individual health more precisely by combining clinical and molecular data from millions of individuals.
About Our Lab
Our research focuses on the development and use of translational and biomedical informatics approaches to address critical challenges in systems medicine and biomedical informatics. We are interested in identifying and developing novel therapeutic and diagnostic approaches for human disease through integration and analysis of molecular and clinical data, and also perform research to develop and evaluate methods to incorporate genomic sequencing data into clinical practice.
Recent Publications
A Top 20 Medical School

